Clínica Cotidiana

Información del artículo
Historia del artículo:
Recibido el 5 de febrero de 2019
Aceptado el 6 de junio de 2019
On-line el 1 de octubre de 2019
Palabras clave:
Enfermedad pulmonar obstructiva crónica
Déficit de alfa-1-antitripsina
Pi*Mmalton
EPOC
Disnea
*Autor para correspondencia
Correo electrónico:
jmherper@hotmail.com
(J.M. Hernández Pérez).
Keywords:
Chronic obstructive pulmonary disease
Alpha-1 antitrypsin deficiency
Pi*Mmalton
COPD
Dyspnea
Beatriz Rescalvo Arjonaa, José María Hernández Pérezb,*, Sergio Fumero Garcíab, Claudia Viviana López Charryc, José Antonio Pérez Pérezd
aCentro de Salud de Breña Alta. La Palma. bSección de Neumología. Hospital General de La Palma. cServicio de Neumología. Hospital Universitario Nuestra Señora de la Candelaria. dInstituto Universitario de Enfermedades Tropicales y Salud Pública de Canarias. Universidad de La Laguna, Área de Genética.
Resumen
La enfermedad pulmonar obstructiva crónica (EPOC) puede producirse por varias causas, la más frecuente el tabaquismo. Sin embargo, hasta el 3 % de los casos son de origen genético, entre ellas la deficiencia de alfa-1-antitripsina (DAAT).
La DAAT es una condición genética, descrita por primera vez en 1963 por Laurell y Eriksson, caracterizada por niveles séricos bajos en sangre de la proteína alfa-1-antitripsina (AAT), lo que predispone al desarrollo temprano de enfisema pulmonar, hepatopatía en niños y adultos y, con menor frecuencia, paniculitis neutrofílica y vasculitis sistémica.
Se han descrito alrededor de 125 variantes alélicas del gen de la AAT, que se clasifican en normales o deficientes; estas a su vez en comunes, raras en función de su frecuencia en la población y las concentraciones séricas que expresan.
A continuación, se presenta el caso clínico de una mujer de 56 años, que debuta con disnea de esfuerzo de 2-3 meses de evolución; tras la realización de diferentes pruebas diagnósticas, se evidencia una DAAT originada por la variante Pi*Z/Mmalton.
© 2019 Sociedad Española de Médicos Generales y de Familia.
Publicado por Ergon Creación, S.A.
Beyond chronic obstructive pulmonary disease
Abstract
Chronic obstructive pulmonary disease can occur for several causes being the most frequent smoking. However, up to 3% of cases are of genetic origin being the alpha-1 antitrypsin deficiency (AATD) the main protagonist.
AATD is a genetic condition, first described in 1963 by Laurell and Erikson, characterized by low serum levels of the alpha-1 antitrypsin (AAT) protein, which predispose to the early development of pulmonary emphysema, hepatopathy in children and adults, and less frequently, neutrophilic panniculitis and systemic vasculitis.
About 125 allelic variants of the gene encoding AAT have been described, which are classified as normal or deficient, and these in turn are common, rare depending on their frequency in the population and the serum concentrations they express.
We present the clinical case of a 56-year-old woman, who debuted with effort dyspnea of 2-3 months of evolution, in which, after performing different diagnostic tests, a DAAT originated by the Pi*Z/Mmalton variant was evidenced.
© 2019 Sociedad Española de Médicos Generales y de Familia.
Published by Ergon Creación, S.A.
Mujer de 56 años de edad, con antecedentes personales de artritis reumatoide y vitíligo, exfumadora de 20 cigarrillos/día desde hace 19 años (IPA 19) y sin alergias conocidas. Acude a la consulta de Neumología remitida desde su centro de salud por disnea de esfuerzo. Refiere que la sintomatología se inició hace 2-3 meses y se presenta ante los moderados-grandes esfuerzos, sin necesidad de pararse cuando caminaba en llano. No refiere tos, ni expectoración, ni fiebre; tampoco manifiesta sintomatología de rinitis, dolor torácico o auto escucha de ruidos respiratorios.
En la exploración física destaca un buen estado general. Se mantiene eupneica en reposo, con una saturación de oxígeno (SpO2) basal de 98 %. Murmullo vesicular conservado sin ruidos respiratorios sobreañadidos. El resto de la exploración es anodina.
La radiografía de tórax no muestra alteraciones significativas.
La espirometría pone en evidencia un patrón obstructivo leve: FVC 2.590 (84 %), FEV1 1.720 (74 %), FEV1/FVC 69,40 %, MEEF 25-75 % 1.130 (53 %) con prueba broncodilatadora negativa.
El hemograma y la bioquímica básica no muestran anormalidades.
Con todo ello se diagnostica en un principio a la paciente de EPOC, fenotipo no agudizador. Se inicia tratamiento con un anticolinérgico de acción larga.
Posteriormente se decide realizar un test de la marcha (Fig. 1). Se objetiva que la paciente recorre 500 m partiendo de una SpO2 basal inicial de 97 %; finaliza la prueba con una SpO2 de 90 %.
Debido a este hallazgo se solicita tomografía axial computarizada de alta resolución (Fig. 2). En ella se confirma la existencia de enfisema principalmente panacinar, predominantemente en las bases.
A partir de estos hallazgos se decide realizar la determinación de los niveles de alfa-1-antitripsina (AAT), cuyo resultado es de 15,1. Se efectúa el genotipado posterior, informado como PiMZ.
Ante la incongruencia de los niveles tan bajos de AAT y el genotipo encontrado, se realiza una secuenciación que confirma la existencia de una variante rara deficitaria, en concreto Pi*Mmalton, es decir, el genotipo de la paciente era en realidad Pi*ZMmalton
Debido a que la paciente presenta criterios para el inicio de tratamiento sustitutivo, tras su aceptación por parte de la misma se inicia dicho tratamiento a una dosis semanal de 60 mg/kg/.
Actualmente la paciente permanece estable y recibe dicho tratamiento.

Figura 1 – Test de la macha a los 6 minutos. Se observa una distancia máxima recorrida de 500 m partiendo de una SpO2 inicial del 97 % terminando en una SpO2 mínima del 90 %.
. Se evidencian signos de enfisema panacinar en ambas bases (flechas rojas).")
Figura 2 – TAC de tórax (ventana pulmonar). Se evidencian signos de enfisema panacinar en ambas bases (flechas rojas).
Comentario
La DAAT es una predisposición genética hereditaria producida por una alteración en el gen SERPINA1 presente en el cromosoma 14, encargado de codificar la AAT1,2. Dicha proteína, sintetizada fundamentalmente en el hepatocito, tiene como función principal inhibir la acción de la elastasa y otras serín-proteasas secretadas por los neutrófilos; actúa así como un potente antiinflamatorio3.
El DAAT confiere predisposición a quienes la portan para el desarrollo temprano de patologías hepáticas como consecuencia del acúmulo intrahepático de los polímeros de la AAT, y para el desarrollo de patología pulmonar tipo enfisema; esto se favorece por las bajas concentraciones plasmáticas y tisulares de AAT, que son insuficientes para la protección del tejido pulmonar de los efectos destructivos de las proteasas sintetizadas ante estímulos irritantes, inflamatorios e infecciosos de la vía aérea2.
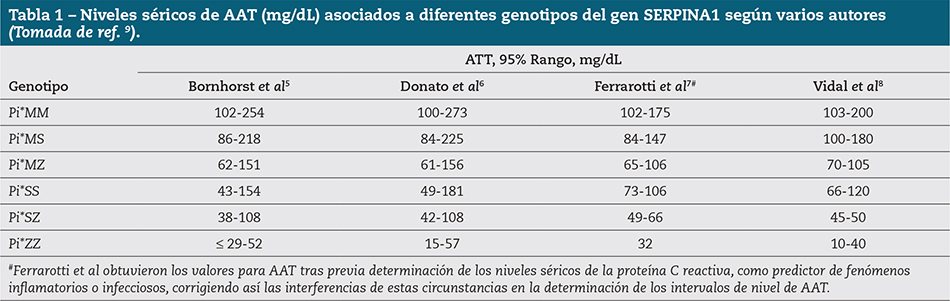
Los alelos normales de la alfa-1-antitripsina se denominan Pi*M, y los deficientes más frecuentes son el Pi*S (A/T, Glu 264Val) y el Pi*Z (G/A, Glu342Lys)2. El déficit grave de la AAT, definido por niveles séricos inferiores al 35% del valor medio establecido (< 50 mg/dl), se asocia en el 95% de los casos al genotipo Pi*ZZ, y el 5% restante se debe a los genotipos Pi*SZ, Pi*MZ o combinaciones de Pi*S o Pi*Z con alelos deficientes nulos o raros4 (Tabla 1)5-9.
En España, la forma grave constituida por la variante Pi*ZZ, tiene una prevalencia de 1 por cada 3.334 habitantes10, pero a pesar de ello es considerada una enfermedad rara. El principal problema es el desconocimiento global de esta patología por muchos médicos de familia y especialistas11, así como que al creer que se trata de una enfermedad rara y poco frecuente, no vale la pena su búsqueda entre los pacientes que acuden a la consulta, dando como resultado un infradiagnóstico importante.
En el caso aquí descrito se encontró una mutación originada por una variante alélica considerada como rara, el alelo Pi*Mmalton (Phe52del)12. La pérdida de la fenilalanina en posición 52 origina un defecto en el plegamiento de la AAT, de la que el 80-90% polimeriza y se acumula en el hígado, expresándose así niveles plasmáticos inferiores al 15% y dando lugar a un incremento del riesgo de desarrollar hepatopatías y enfisema pulmonar13. En la población española, según el Registro Español de pacientes con DAAT (REDAAT), los alelos raros representan un 4,6% de las variantes deletéreas registradas, siendo la variante Pi*Mmalton un de las más frecuentemente observada en la población española4.
Este infradiagnóstico, y la gran prevalencia de los alelos deficitarios en nuestro entorno, hacen necesario un buen diagnóstico de esta patología ante la más mínima sospecha clínica, radicando la importancia de la detección temprana en la promoción del cese tabáquico, tratamiento precoz del enfisema, así como el estudio genético familiar correspondiente y tratamiento precoz sustitutivo en aquellos casos indicados (como la paciente descrita en el presente caso clínico), ya que la instauración de este aumenta su supervivencia disminuyendo la pérdida de densidad pulmonar14.
Para la confirmación diagnóstica del DAAT se debe realizar una determinación sérica de los niveles de AAT y continuar el diagnóstico con la determinación del genotipo (prueba de referencia), si bien en algunos, todavía se suele utilizar el fenotipo15,16. Si nos encontramos una situación (como el presente caso) donde los niveles detectados de la AAT discrepan con el genotipo en cuanto a la presencia de las variantes genéticas no-S/S y no-Z/Z, es necesario llegar a la secuenciación del gen SERPINA1, prueba referente para la identificación de mutaciones especificas asociadas a alelos raros nulos o deficitarios17 (Fig. 3).
.")
Figura 3 – Algoritmo para el diagnóstico de la DAAT. (Tomada de ref. 16).
Además, la Organización Mundial de la Salud18 y la Sociedad Española de Neumología y Cirugía Torácica15 recomiendan que en todos los individuos diagnosticados de EPOC se deben analizar para el DAAT, al menos una vez en la vida, mediante cuantificación de los valores séricos. De igual manera, según los criterios SEPAR, se recomienda testar también a los individuos indicados en la tabla 2.
Como conclusión, ante cualquier paciente con diagnóstico de EPOC debemos solicitar los niveles de AAT para descartar la DAAT y evitar que un paciente, que de otra manera podría beneficiarse de una terapia específica, pase inadvertido ante nuestros ojos. La DAAT no es una enfermedad rara, sino desgraciadamente raramente diagnosticada.
.")
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.
Bibliografía
- American Thoracic Society/European Respiratory Society statement. Standards for the diagnosis and management of individuals with alpha1antitrypsin deficiency. Am J Respir Crit Care Med. 2003; 168: 818-900.
- Blanco I, Lara B. Déficit de alfa-1-antitripsina: fisiopatología, enfermedades relacionadas y tratamiento. Barcelona: RESPIRA; 2016. p. 327.
- Blanco I. Blanco’s Overview of alpha1antitrypsin deficiency: History, biology, pathophysiology, related diseases, diagnosis, and treatment. Elsevier-Academic Press; 2017.
- Lara B, Blanco I, Teresa M, Rodríguez E, Bustamante A, Casas F, et al. Spanish Registry of patients with alpha1antitrypsin deficiency: Database evaluation and population analysis. Arch Bronconeumol. 2017; 53: 13-8.
- Bornhorst JA, Greene DN, Ashwood ER, Grenache DG. Alpha1antitrypsin phenotypes and associated serum protein concentrations in a large clinical population. Chest. 2013; 143: 1000-8.
- Donato LJ, Jenkins SM, Smith C, Katzmann JA, Snyder MR. Reference and interpretive ranges for alpha1antitrypsin quantitation by phenotype in adult and pediatric populations. Am J Clin Pathol. 2012; 138: 398-405.
- Ferrarotti I, Thun GA, Probst-Hensch NM, Luisetti M. Alpha1antitrypsin level and pheno/genotypes. Chest. 2013; 144: 1732-3.
- Vidal R, Blanco I, Casas F, Jardí R. Diagnóstico y tratamiento del déficit de alfa-1-antitripsina. Arch Bronconeumol. 2006; 42: 645-59.
- Hernández Pérez JM. Estudio de la prevalencia de la deficiencia de alfa-1-antitripsina en la isla de La Palma. Tesis Doctoral. Universidad de La Laguna; 2017.
- Blanco I, Bueno P, Diego I, Pérez-Holanda S, Casas-Maldonado F, Esquinas C, et al. Alpha1antitrypsin Pi*Z gene frequency and Pi*ZZ genotype numbers worldwide: an update. Int J Chron Obstruct Pulmon Dis. 2017; 12: 561-9.
- Greulich T, Ottaviani S, Bals R, Lepper PM, Vogelmeier C, Luisetti M, et al. Alpha1antitrypsin deficiency: Diagnostic testing and disease awareness in Germany and Italy. Respir Med. 2013; 107: 1400-8.
- Belmonte I, Barrecheguren M, López-Martínez RM, Esquinas C, Rodríguez E, Miravitlles M, et al. Application of a diagnostic algorithm for the rare deficient variant Mmalton of alpha1antitrypsin deficiency: A new approach. Int J COPD. 2016; 11: 2535-41.
- Curiel DT, Holmes MD, Okayama H, Brantly ML, Vogelmeier C, Travis WD, et al. Molecular basis of the liver and lung disease associated with the alpha1antitrypsin deficiency allele Mmalton. J Biol Chem. 1989; 264: 13938-45.
- The alpha1antitryspin deficiency registry study group. Survival and FEV 1 decline in individuals with severe deficiency of alpha-1 -Antitrypsin. Crit Care Med. 1998; 158: 49-59.
- Casas F, Blanco I, Martínez MT, Bustamante A, Miravitlles M, Cadenas S, et al. Actualización sobre indicaciones de búsqueda activa de casos y tratamiento con alfa-1-antitripsina por vía intravenosa en pacientes con enfermedad pulmonar obstructiva crónica asociada a déficit de alfa-1-antitripsina. Arch Bronconeumol. 2015; 51: 185-92.
- Lopes AP, Mineiro MA, Costa F, Gomes J, Santos C, Antunes C, et al. Portuguese consensus document for the management of alpha1antitrypsin deficiency. Pulmonology. 2018; 24: 1-21.
- Hernández Pérez JM, Ramos Díaz R, Fumero García S, Pérez Pérez JA. Description of alpha1antitrypsin deficiency associated with PI*Q0ourém allele in La Palma Island (Spain) and a genotyping assay for detection. Arch Bronconeumol. 2015; 51: e13.
- Anonymous. Alpha1antitrypsin deficiency: memorandum from a WHO meeting. Bull WHO. 1997; 75: 397-415.